Getting started#
Installation#
dcmri can be installed using pip:
pip install dcmri
Basic usage#
The most common application of DCMRI involves measurement of the endothelial
permeability using a steady-state MRI sequence
and a Tofts model. In this
section we illustrate the basic usage of dcmri by applying this model to
synthetic data.
We start by import the package and generating synthetic region-of-interest
(ROI) data using dcmri.fake_tissue.
We want these to look realistic, so we are adding noise with a
contrast-to-noise ratio (CNR) of 50:
import dcmri as dc
time, aif, roi, _ = dc.fake_tissue(CNR=50)
Here time is an array of time points, aif is a signal-time curve measured in a feeding artery at those times, and roi is a signal-time curve measured in a region of interest.
Next we find a suitable tissue type from the
tissue bank and initialize it. For most common
applications, this will be dcmri.Tissue:
tissue = dc.Tissue(aif=aif, t=time)
At this point this is still a generic tissue with default parameter values. The next step is to train the tissue using the ROI data:
tissue.train(time, roi)
And that’s it. We can now display the measured tissue parameters:
tissue.print_params(round_to=3)
--------------------------------
Free parameters with their stdev
--------------------------------
Blood volume (vb): 0.018 (0.002) mL/cm3
Interstitial volume (vi): 0.174 (0.004) mL/cm3
Permeability-surface area product (PS): 0.002 (0.0) mL/sec/cm3
----------------------------
Fixed and derived parameters
----------------------------
Tissue Hematocrit (H): 0.45
Plasma volume (vp): 0.01 mL/cm3
Interstitial mean transit time (Ti): 74.614 sec
B1-corrected Flip Angle (FAcorr): 15 deg
The standard deviations of the free parameters are orders of magnitude smaller than the value itself, which offers confidence that the tissue properties are well determined by the data.
We should also verify that the trained tissue does indeed predict the data correctly:
tissue.plot(time, roi)
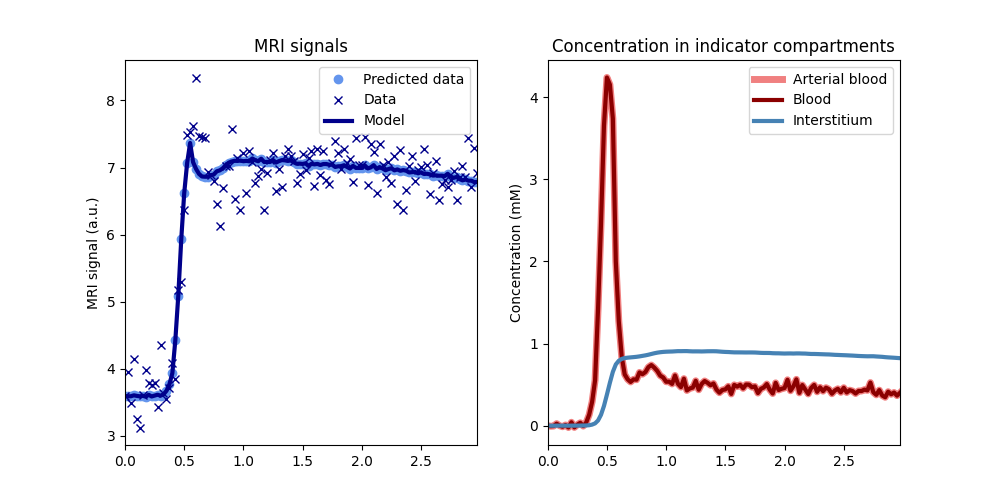
The signal plot on the left shows that the model correctly predicts the measured data, except for the noise. The plot on the right shows that the reconstructed concentrations in blood and tissue have the expected profiles and that values are in an expected range for a standard contrast agent injection (0-5mM in blood).
Customizing the tissue model#
Since the extended Tofts model is the most widely used approach, it is set as
default in dcmri.Tissue and does not need to be specified explicitly.
Similarly other configurations such as initial values, parameter ranges and
which parameters to fix in the analysis - are set to reasonable defaults.
Analysing the data with other configurations is not much more difficult. Let’s do the analysis again, but this time allowing for restricted water-exchange across the tissue cell walls. We can do this by setting the water exchange model to ‘FR’, meaning fast (F) water exchange across the endothelium, and restricted (R) water exchange across the tissue cell walls:
tissue = dc.Tissue(aif=aif, t=time, water_exchange='FR')
tissue.train(time, roi).print_params(round_to=3)
--------------------------------
Free parameters with their stdev
--------------------------------
Transcytolemmal water PS (PSc): 533.268 (19598.606) mL/sec/cm3
Blood volume (vb): 0.018 (0.002) mL/cm3
Interstitial volume (vi): 0.174 (0.007) mL/cm3
Permeability-surface area product (PS): 0.002 (0.0) mL/sec/cm3
----------------------------
Fixed and derived parameters
----------------------------
Tissue Hematocrit (H): 0.45
Plasma volume (vp): 0.01 mL/cm3
Interstitial mean transit time (Ti): 74.605 sec
Intracellular water mean transit time (Twc): 0.002 sec
B1-corrected Flip Angle (FAcorr): 15 deg
We now get an additional value for the water exchange exchange across the cell wall - which is very high in this case (and imprecise) because the synthetic data are generated for a tissue with infinite water exchange.
Any other options can be set in the same way. As an example, we run this same model again, but this time we provide a different initial value for the blood volume, and we treat the B1-correction factor as a free parameter with bounds [0,2] (by default this is fixed to 1):
tissue = dc.Tissue(aif=aif, t=time, water_exchange='FR', vb=0.5)
tissue.set_free(B1corr=[0,2])
Train the tissue again and print the parameters. We illustrate here also how methods in dcmri tissue models can be chained:
tissue.train(time, roi).print_params(round_to=3)
--------------------------------
Free parameters with their stdev
--------------------------------
Transcytolemmal water PS (PSc): 264.663 (16140.959) mL/sec/cm3
Blood volume (vb): 0.017 (0.003) mL/cm3
Interstitial volume (vi): 0.169 (0.022) mL/cm3
Permeability-surface area product (PS): 0.002 (0.0) mL/sec/cm3
Tissue B1-correction factor (B1corr): 0.98 (0.027)
----------------------------
Fixed and derived parameters
----------------------------
Tissue Hematocrit (H): 0.45
Plasma volume (vp): 0.009 mL/cm3
Interstitial mean transit time (Ti): 74.373 sec
Intracellular water mean transit time (Twc): 0.003 sec
B1-corrected Flip Angle (FAcorr): 14.701 deg
Since the synthetic data are generated with an exact flip angle, the B1 correction is close to 1 - as expected.
Pixel-based analysis#
dcmri includes dedicated tools for pixel-based analysis. To illustrate these,
we analyse synthetic brain images with an extended Tofts model.
First let’s generate some synthetic brain images. For the purpose of this illustration we use coarse 128 x 128 images:
n = 128
time, signal, aif, _ = dc.fake_brain(n)
In this case, time and aif are still 1D arrays of time points, but signal is 3D array (2D + time) with pixel-based synthetic data.
Since the data are now images, we analyse them using dcmri.TissueArray
instead of dcmri.Tissue. Since pixel-based computations take more time, we
display a progress bar during computations by setting the verbosity to 1:
shape = (n, n)
image = dc.TissueArray(shape, aif=aif, t=time, verbose=1)
Now we train the image model on the data and plot the results:
image.train(time, signal)
image.plot(time, signal)
As before - by default this runs the extended Tofts model for every pixel. The model predicts the data well (left) but parameter maps (right) show unrealistic PS values in the normal brain tissue - a known issue with this model in the brain. Let’s analyse these data again with a model that is more suitable for whole-brain analysis (2-compartment uptake model, aka 2CU):
image = dc.TissueArray(shape, aif=aif, t=time, verbose=1, kinetics='2CU')
Train the model on the data and plot the maps again:
image.train(time, signal).plot(time, signal)
The PS values are now zero everywhere, except in the lesions with broken blood-brain barrier. The maps in the lower row also show that parameter estimates are unreliable in the ventricles, which are not accessible to the contrast agent.
Further customization of pixel-based models, such as
setting initial values or modifying parameter ranges, also works in the same
way as for the ROI-based model dcmri.Tissue.